Purpose:
Make RFP proteins (extrated from jelly fish) in bacteria and to learn the steps required to genetically engineer.
Procedure and Materials:
Lab 2a:
Procedure:
- Label two empty microfuge tubes, "R+" and "R-" using a permanent marker
- Add 4.0 ul of 2.5xb, 4.0 pR, and 2.0 ul to the "R+" Tube
- Add 4.0 ul of 2.5xb, 4.0 pR, and 2.0 ul of Distilled Water to the "R-" Tube
- Centrifuge both tubes for four seconds
- Place both tubes into Water Bath and incubate for at least an hour, then remove both tubes and place in a freezer at -20 degrees Celsius
Materials:
|
|
Lab 4a:
Procedure:
- Add 2.0 ul of loading dye to both the "R+" and "R-" tubes
- Microfuge both tubes for several seconds
- Fill electrophoresis box with 1xSB to cover gels
- Dispense 10.0 ul of loading dye, 10.0 ul of "R-", and 10.0 ul of "R+" in individual wells and note their position
- Put lid over box
- Connect the corresponding wires, turn the power on, and set the voltage to 130-135 volts
- After 45-50 minutes, remove gel and examine/photograph
Materials:
|
|
Lab 5a:
Procedure:
- Label 2 empty tubes "P+" and "P-"
- Add 50 ul of competent cells to each tube and keep on ice for remainder of lab
- Add 10 ul of RP to "P+" tube and mix using pipette
- Keep on ice for 15 minutes
- Label each petri dish either LB (Divide in half), LB/Amp (Divide in half), or LB/Amp/Ara (leave whole)
- Carry "P+" and "P-" tubes to 42 degrees Celsius water bath and place them for exactly 45 seconds
- Place back on ice for 1-2 minutes
- Add 150 ul of LB to each tube and gently flick to mix
- Let sit at room temperature for 15 minutes
- Pipet 50 ul of "P-" cells to half of the LB and LB/Amp dishes
- Spread cells using steile spreader
- Repeat for "P+" cells
- Allow dishes to sit right-side up for five minutes and then tape together
- Flip upside-down and let rest in 37 degrees Celsius water bath and incubate for 24-36 hours
Materials:
|
|
Lab 6: Part A
Procedure:
- Record color of EC tube
- Spin EC tube in microcentrifuge for 5 minutes
- Remove 200 ul of supernatant from EC tube
- Add 1,000 ul of LB/Amp/Ara E. Coli to EC tube
- Repeat steps 2 and 3
- Remove as much supernatant from EC tube as possible
- Add 150 ul of EB to EC tube
- Drag tube across tube rack to resuspend cells
- Add 150 ul of LyB to EC tube and resuspend
- Incubate cells at room temperature overnight
- Dispose of used tips and tubes
Materials:
|
|
Lab 6: Part B
Procedure:
- Label 2 clean microfuge tubes "SUPER" and "RFP"
- Set liquid waste container underneath stopcock
- Open column and allow liquid to drain into waste container
- Close valve when about 1-2 mm of liquid are left above resin bed
- Microcentrifuge EC tube for about 5 minutes
- Using the P-1000 micropipette, remove 200 ul of supernatant and dispense into "SUPER" tube
- Add 200 ul of BB to the "SUPER" tube and mix gently
- Add 400 ul of "SUPER" tube to the chromatography tube
- Open valve and allow column to drain, but close the valve when 1-2mm of liquid is left above resin bed
- Add 1,000 ul of WB down the side of the column
- Open the valve and allow the column to drain, but close valve when 1-2mm of liquid is left above resin bed.
- Add 2,000 ul of EB down the side of the column
- Set the "RFP" tube under stopcock and allow only the red substance into the tube
- Add 2,000 ul of CEB to column and cap tightly
- Pour waste collection down the drain
- Compare color between other groups
Materials:
|
|
EXPERIMENTAL Overview:
Lab 2a:
Verification of a plasmid by restriction digest
-Cut plasmid with BanHI and HindIII to cut out RFP-ara from bacteria plasmid
-Cut plasmid with BanHI and HindIII to cut out RFP-ara from bacteria plasmid
Lab 4a:
Verification of plasma digest by gel electrophoresis
Lab 5a:
Transformation of bacteria with recombinant plasmid
Lab 6:
Portification of RFP using chromatography
Data:
Lab 2a:
Before the Lab:
1. If pARA-R is digested with BamHI and HindIII, what fragments are produced? Record the nucleotide sequence of the sticky ends and the length of each fragment (bp), and indicate the genes and other important sequences present on each fragment.
The two fragments produced are an RFP fragment with pBAD and an Ara-C fragment with ori and Amp-R. The nucleotide sequence length of the RFP fragment is 807 BP and the Ara-C fragment is 4495 BP.
The two fragments produced are an RFP fragment with pBAD and an Ara-C fragment with ori and Amp-R. The nucleotide sequence length of the RFP fragment is 807 BP and the Ara-C fragment is 4495 BP.
2. In order to create a plasmid that can produce the red fluorescent protein in bacteria, what components are needed in the plasmid?
To produce the red fluorescent protien in our bacteria, the RFP gene as well as Ara-C (which binds to the promoter region) are needed.
To produce the red fluorescent protien in our bacteria, the RFP gene as well as Ara-C (which binds to the promoter region) are needed.
3. Bacteria can be killed by an antibiotic unless they carry a plasmid that has the gene for resistance to that antibiotic. Biotechnologists call these genes selectable markers because only bacteria that carry the gene will survive an antibiotic. If the uptake of DNA by bacteria is inefficient, why is a selectable marker critical in cloning a gene in bacteria?
A selectable marker is extremely important to cloning genes in bacteria because it determines which bacteria will continue to grow, while the bacteria without the marker die, thus separating the two colonies.
A selectable marker is extremely important to cloning genes in bacteria because it determines which bacteria will continue to grow, while the bacteria without the marker die, thus separating the two colonies.
Lab 2a Questions:
1. List in words or indicate in a drawing the important features of a plasmid vector that are required to clone a gene. Explain the purpose of each feature.
Plasmid Vector:
Plasmid Vector:
- Ori: Origin of replication
- RFP and PBad: Gene of interest
- Amp-R: Selective marker
- Ara-C: Binds to the promoter region which leads to transcription
2. What role do restriction enzymes have in nature?
Restricion enzymes act as a natural defense against bacteria. They cut out DNA of other bacteria (killing them) to help their own chances of survival.
Restricion enzymes act as a natural defense against bacteria. They cut out DNA of other bacteria (killing them) to help their own chances of survival.
3. Using your understanding of evolution, why would bacteria retain a gene that gives them resistance to antibiotics? How is the existence of bacteria with antibiotic resistance affecting medicine today?
Antibiotic resistance genes would be kept because bacteria could survive in the presence of what could be lethal. These resistant bacteria affect medicine because stronger antibiotics are needed for less severe bacteria as well as new methods of killing bacteria needing to be developed for those strong bacteria.
Antibiotic resistance genes would be kept because bacteria could survive in the presence of what could be lethal. These resistant bacteria affect medicine because stronger antibiotics are needed for less severe bacteria as well as new methods of killing bacteria needing to be developed for those strong bacteria.
4. Bacteria, sea anemones, and humans seem, on the surface, to be very different organisms. Explain how a gene from humans or a sea anemone can be expressed in bacteria to make a product never before made in bacteria.
All genes have the same central dogma (DNA -> mRNA ->Protein), so they can be expressed in seemingly different organisms.
All genes have the same central dogma (DNA -> mRNA ->Protein), so they can be expressed in seemingly different organisms.
5. Due to a mishap in the lab, bacteria carrying a plasmid with an ampicillinresistant gene and bacteria carrying a plasmid with a gene that provides resistance to another antibiotic (kanamycin) were accidentally mixed together. Design an experiment that will allow you to sort out the two kinds of bacteria.
Put half of the colony in a petri dish of ampacillin and the other half into a dish of kanamycin. The ampacillin dish will be left with only ampacillin-resistant bacteria and the kanamycin dish will only have kanamycin-resistant bacteria.
Put half of the colony in a petri dish of ampacillin and the other half into a dish of kanamycin. The ampacillin dish will be left with only ampacillin-resistant bacteria and the kanamycin dish will only have kanamycin-resistant bacteria.
Lab 4a:
Before the Lab:
1. The pARA-R plasmid you digested in Laboratory 2A was replicated in a bacterial cell. What configurations—supercoiled, nicked circle, and multimer—might the plasmid have before digestion?
The plasmid can have all three configurations: supercoiled, nicked circle, and multimer.
The plasmid can have all three configurations: supercoiled, nicked circle, and multimer.
2. You need to catalog all the products you might see, including the different plasmid configurations. Review your work in Laboratory 2A. What products might you expect to see in the R– and R+ tubes? Create a table that shows all the possible fragments and plasmids by tube. Include the length (bp size) of each possible fragment or plasmid, and arrange the products found in each microfuge tube by size, from smallest to largest. Include any possible plasmid configurations, and arrange them first by size and next by speed through the gel, from fastest to slowest.
|
Tube
R+
|
Fragments (in order of increasing BP size)
1. pARA-R: 5,302 BP
Can have all three combinations Supercoiled configuration = fastest |
|
R-
|
1. RFP-pBAD: 807 BP
2. Amp-R,Ori, Ara-C: 4,495 BP |
3. Read through the Methods section on pages 64 through 66 and briefly outline the steps, using words and a flowchart.
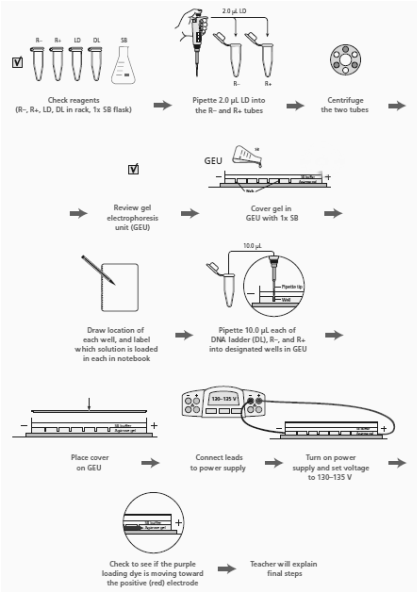
Lab 4a Questions:
1. Why is it important to verify that you have the correct recombinant plasmid?
It is important to verify because during the lab, we could make measuring mistakes, the materials could be faulty, or make errors in our procedure.
It is important to verify because during the lab, we could make measuring mistakes, the materials could be faulty, or make errors in our procedure.
2. How did your actual gel results compare to your gel predictions?

Our R+ and R- columns came out perfectly, however our column for loading dye did not show up at all.
3. Do you see any bands that are not expected? What could explain the origin of these unexpected bands?
Our group did not have any unexpected bands, we only had unexpected blank gel space.
Our group did not have any unexpected bands, we only had unexpected blank gel space.
4. Does the gel photograph show that you are using the correct recombinant plasmid? Describe the evidence you used to make this assessment.
This photo shows that we are using the correct recombinant plasmid--the R+ lane has two fragments in the correct spots.
This photo shows that we are using the correct recombinant plasmid--the R+ lane has two fragments in the correct spots.
5. In the R- lane, do you see evidence of multiple configurations of plasmids? Explain your answer.
In the R- lane, two different bands prove multiple plasmid configurations.
In the R- lane, two different bands prove multiple plasmid configurations.
6. In the R+ lane, do you see evidence of complete digestion? Explain your answer.
The fact that the land only has two fragments shows that the plasmid was completely digested into its two fragments.
The fact that the land only has two fragments shows that the plasmid was completely digested into its two fragments.
7. Where would you expect to see the RFP gene and the Amp-R gene in the gel photograph? Are you able to locate these two genes? Explain your answer.
In the R+ lane, we expect to see a band around 807 bp as well as a band at around 4,495 bp. We were able to locate these two genes as evident in our picture.
In the R+ lane, we expect to see a band around 807 bp as well as a band at around 4,495 bp. We were able to locate these two genes as evident in our picture.
8. Compare the lanes that have linear fragments with the lanes that have plasmids. Is there a difference in the shape of the bands between these two DNA forms?
The bands for the linear fragments have trailing edges instead of the non-trailing edges of the lanes that have plasmids.
The bands for the linear fragments have trailing edges instead of the non-trailing edges of the lanes that have plasmids.
Lab 5a:
Before the Lab:
1. Ampicillin is an antibiotic that kills bacterial cells by disrupting the formation of cell walls. However, the pARA-R plasmid has the ampicillin resistance gene, which produces a protein that breaks down ampicillin. What is the purpose of growing bacteria that have been transformed in the presence of ampicillin?
The purpose of growing these bacteria is for the purpose of seperation between those with the resistant gene and those without.
The purpose of growing these bacteria is for the purpose of seperation between those with the resistant gene and those without.
2. What will happen when bacterial cells that contain the pARA-R plasmid are not given arabinose?
When they are without arabinose the RFP gene will not be expressed -- no red glow.
When they are without arabinose the RFP gene will not be expressed -- no red glow.
3. In the lab, you will add samples of the control group P– and the treatment group P+ to plates that contain various combinations of Luria Broth (LB), ampicillin, and the sugar arabinose. Predict the growth on each dish.
I predict that Plate I (LB) will have non-glowing growth on both sides. I predict that Plate II (LB/Amp) will have non-glowing growth on the P+ side. For Plate III (LB/Amp/Ara), I predict a glowing red colony.
I predict that Plate I (LB) will have non-glowing growth on both sides. I predict that Plate II (LB/Amp) will have non-glowing growth on the P+ side. For Plate III (LB/Amp/Ara), I predict a glowing red colony.
Lab 5a Questions:
1. Look at the results of your transformation. Do your actual results match your predicted results? If not, what differences do you see, and what are some explanations for these differences?
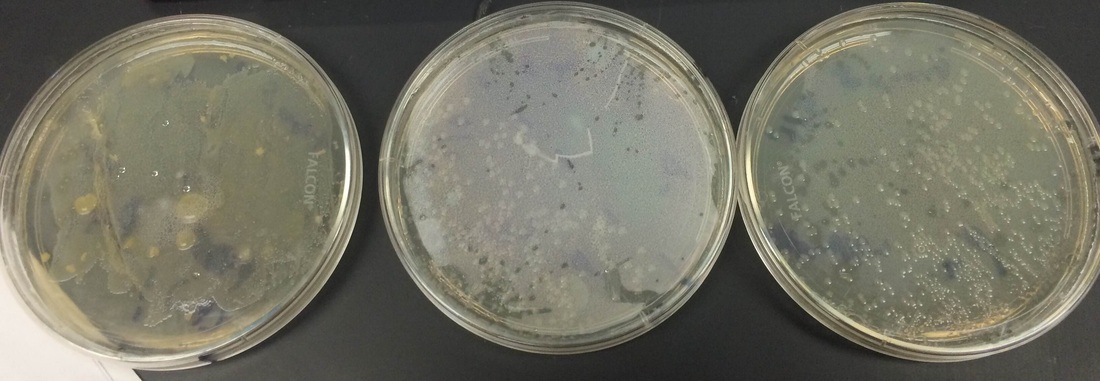
Our results mostly match our predictions. Unfortunately, the colony on the LB/Amp/Ara plate was not glowing red as it should have. This was not our fault, but rather the company shipped us faulty materials.
|
|
2. How many red colonies were present on your LB/amp/ara plate?
There were no red colonies present on our plate.
There were no red colonies present on our plate.
3. Why did the red colonies only appear on the LB/Amp/Ara plate and not the LB/Amp plate?
The red colonies would have only appeared on the plate with araganose because araganose allows the red protein to be expressed.
The red colonies would have only appeared on the plate with araganose because araganose allows the red protein to be expressed.
4. Recombinant plasmids are engineered so that they can replicate in the cell independently of the chromosome replication. Why is it important to have multiple copies of a recombinant plasmid within a cell?
There are multiple copies of the recombinant plasmid because the more copies, the more product produced within the cell.
There are multiple copies of the recombinant plasmid because the more copies, the more product produced within the cell.
5. How is the information encoded in the RFP gene expressed as a trait? Be sure to use what you have previously learned about gene expression and the relationship between DNA, RNA, protein, and traits.
The central dogma (DNA ->RNA->Protein) ends with a protien. The RFP protein is expressed in the form of the red glow as a trait.
The central dogma (DNA ->RNA->Protein) ends with a protien. The RFP protein is expressed in the form of the red glow as a trait.
6. Why is it possible for bacteria to make a human protein, such as insulin, or a sea anemone protein, such as the red fluorescent dye?
All genes have the same central dogma (DNA -> mRNA ->Protein), so they can be expressed in seemingly different organisms.
(See Lab 2a: Question 4)
All genes have the same central dogma (DNA -> mRNA ->Protein), so they can be expressed in seemingly different organisms.
(See Lab 2a: Question 4)
Lab 6:
Before the Lab:
1. How can solutions of different salt concentrations, which will unfold proteins to varying degrees, be used to help purify red fluorescent protein using column chromatography?
Proteins are unfolded in the highly salt concentrated buffer. The proteins that don't flow out the coulmn are refolded when lower salt concentrations are added, thus they leave the column.
Proteins are unfolded in the highly salt concentrated buffer. The proteins that don't flow out the coulmn are refolded when lower salt concentrations are added, thus they leave the column.
Lab 6 Questions:
1. Why is a protein’s conformation important for carrying out its function?
A protein's conformation is important because it determines the promoter regions of the given protein. These promoter regions are what give each protein their different functions.
A protein's conformation is important because it determines the promoter regions of the given protein. These promoter regions are what give each protein their different functions.
2. What properties of the amino acids in a protein relate to protein folding?
The sequence of an amino acid determines how it will fold into a protein.
The sequence of an amino acid determines how it will fold into a protein.
3. Does the eluate containing your red fluorescent protein appear less bright or brighter than it did in the cell lysate following centrifugation? If there is a noticeable difference in the intensity of the red color, what might account for that?
The RFP-containg elute is brighter than the cell lysate. This is because the elute contains the most RFP--the cell lysate contained all the cell proteins.
The RFP-containg elute is brighter than the cell lysate. This is because the elute contains the most RFP--the cell lysate contained all the cell proteins.
4. What characteristic of red fluorescent protein is used as the basis for separation by column chromatography?
The red fluorescent protein sticks to the resin column when unfolded (more hydrophobic amino acids).
The red fluorescent protein sticks to the resin column when unfolded (more hydrophobic amino acids).
5. How might the column chromatography procedure be adjusted or modified to increase the purity of the red fluorescent protein sample?
If we repeated the process with more wash buffers and were more careful about collection, we could increase the red fluorescent protein purity.
If we repeated the process with more wash buffers and were more careful about collection, we could increase the red fluorescent protein purity.
Analysis/Conclusion:
In this lab, we attempted to make a glowing red bacteria colony. While they might not have been glowing, we did manage to grow a colony with the correct gene (red fluorescent protein). We did this by using restriction enzymes to isolate the RFP gene and then confirmed that our work was correct by using gel electrophoresis. Then we used recombinant plasmids and chromotography to seperate the red protein into a tube.
After extracting the RFP, we ran it in a gel:

Our particular column was the purple line on the first gel. We had very little protein in the proper places and our gel had some issues as seen in the odd formation. however when looking at 2nd and 3rd period's gels. We can see that some groups had really good RFP extraction. They had a nice thick band at the proper place according to the chart above.
Reflection:
This lab was very long and confusing, but the was the first lab that our teamwork got better as it progressed. Unlike other labs which started off effective and went downhill into utter chaos, this lab went from a decent group to a functioning one. This lab felt very rushed during some parts and very drawn out in others, and the division of time due to the Forensics project made it challenging to compile all the information mentally. Throughout this lab, my pipetting skills have gotten considerably better and I am now confident in my ability to do most lab tasks. However, I struggled with many tasks that could have been easy, but were difficult due to poor communication. Overall, this lab was a useful one, but could have been a lot more simple and more effective from a learning standpoint.
